TR4
TR4 is a highly selective β2-adrenoceptor antagonist (β2-AR blocker), unlike the currently available β-AR blockers which are either selective for the β1-AR or non-selective (block β1-AR and β2-AR). In recent years, pre-clinical studies have shown that signalling via the β2-AR is far more complicated than previously thought, and that a selective β2-AR blocker, such as TR4, may be a potential new treatment for chronic heart failure, chronic asthma and pancreatic cancer.
Chronic heart failure
Patients with chronic heart failure (CHF) have reduced cardiac output, fluid retention and raised venous pressure1. Cardiac contraction and relaxation are controlled by the β1-AR and β2-AR on cardiomyocytes, via G-protein coupled receptor (GPCR) pathways. Unlike the β1‑AR, which couples exclusively to a stimulatory Gαs pathway, the β2-AR can couple to both a stimulatory Gαs (classical) pathway and an inhibitory Gαi (non-classical) pathway (Figure 1). Both β1- and β2-AR exert their positive inotropic actions through the “classical” pathway, by coupling to Gαs, which leads to an increase in cAMP and activation of protein kinase A (PKA). However, in animal models of heart failure, the ratio of β1/β2-AR density in the normal heart is reversed, and phosphorylation of the β2-AR receptor switches it from Gαs coupling to the Gαi pathway2. Gαi coupling also activates other pathways that are thought to be harmful (Figure 1):
- the phosphodiesterase 4 (PDE4) pathway, which blocks cAMP production;
- the phosphoinositide 3-kinase (PI3K) pathway, which leads to an increase of inflammatory cytokines; and
- the Ras/Raf/mitogen-activated protein kinase (MAPK) and ERK kinase signalling pathways, which are involved in cell proliferation, apoptosis and left ventricular hypertrophy.
Figure 1. Mechanism of classical vs non-classical signalling of the β-AR

AC, adenylate cyclase; Akt, protein kinase B; arrestin, β-arrestin; β1-AR, β1-adrenoceptor; β2-AR, β2-adrenoceptor; cAMP, cyclic adenosine monophosphate; ERK, extracellular regulated kinase; Gαs, heterotrimeric G-protein alpha subunit; Gαi, heterotrimeric G-protein inhibitory subunit; GRb2, growth factor receptor-bound protein 2; GRK, G-protein receptor kinase; MAPK , mitogen-activated protein kinase; MEK, mitogen activated protein kinase/ERK kinase; NFkB, nuclear factor kappa B subunit; PDE IV/3B, phosphodiesterase IV/3B; PI3K, phosphoinositide 3-kinase; PKA, protein kinase A; Raf, rapidly accelerated fibrosarcoma kinase; Ras, Ras family protein; Shc, SHC-transforming protein 1; Sos, Son of Sevenless protein; P, phosphorylation.
When propranolol, the first widely used β-blocker, was introduced in the late 1960s, it carried a dual warning against its use in patients with heart failure or asthma because it had been found to worsen those conditions. Meanwhile, β-AR had been shown to be of two types – β1 found mainly in the heart, and β2 found mainly in the bronchi3. Later, many other β-AR antagonists with varying degrees of selectivity were discovered. For example, metoprolol and bisoprolol are selective β1-AR antagonists, ICI 118,551 is a selective β2-AR antagonist4, and carvedilol and bucindolol are non-selective antagonists – they antagonise both β1 and β2-AR5.
However, contrary to the prevailing belief, Waagstein et al 19756 showed that patients with tachycardia from CHF improved after treatment with a β-blocker. They gave patients a very small starting dose and increased it gradually over 2–12 months until an effective dose was reached. After a hiatus of almost 20 years, major clinical trials were eventually done to assess the effectiveness of β-blockers metoprolol, bisoprolol, carvedilol and bucindolol in CHF7. All those trials used a dose-titration regimen.
Several β-AR blockers, such as carvedilol and metoprolol, are now licensed to treat CHF. Although they reduced mortality in clinical trials, they are underused in patients with CHF in clinical practice because of the need for the complicated and time-consuming dose-titration regimen8.
At the start of the 21st century, based on experiments with mice engineered to overexpress the human β2-AR by 200-fold, the late Sir James Black, who invented propanolol, proposed that, contrary to conventional belief, a selective β2-AR antagonist is a more appropriate treatment of CHF9. Current β-AR antagonists all antagonise the β2-AR, at least to some degree. Despite the prevailing belief that β1-AR antagonism is necessary to treat CHF, subsequent studies showed that none of the approved β-ARs were in fact highly selective for the β1-AR and, at a clinical dose, they all have significant β2-AR blocker effects5,10. Thus, β2-AR antagonism may be responsible for the therapeutic effect.
We plan to test James Black’s theory that the ideal β-AR blocker to treat CHF is one that’s selective for the β2-AR, such as TR4, and avoids the need for dose titration1. Dose-titration of currently used β-AR antagonists is required because acute administration has the unwanted effect of antagonising the β1-AR, thereby blocking signalling through a receptor that promotes cardiac contraction. James Black proposed that β1-AR activity in the failing heart increases during dose-titration such that the therapeutic dose of the β-AR antagonist does not significantly inhibit signalling mediated by the β1-AR population through Gαs, but does inhibit the deleterious effects mediated by β2-AR signalling through Gαi. Consequently, by the time the therapeutic dose is reached, after several weeks or months of dose-titration, there is enough β1-AR activity in the heart to counter the β1-AR blockade and thus to maintain cardiac function. Because β2-AR signalling through Gαi is also inhibited, chronic dosing improves cardiac function.
That theory would also explain why the non-selective β-blocker carvedilol was preferred over the β1-AR selective metoprolol for the treatment of CHF in the COMET trial11. Carvedilol is in fact a weakly selective β2-AR antagonist, whereas metoprolol is a weakly selective β1-AR antagonist5. Thus, for any given dose, carvedilol exhibits a proportionally higher, beneficial β2-AR antagonism than metoprolol.
In conclusion, TR4 could eliminate the need for dose-titration, inhibit signalling through the Gαi coupled β2-AR, and avoid the unwanted effects of inhibiting β1-AR mediated cardiac contraction.
Chronic asthma
Asthma is a respiratory condition that affects 5.4 million people in the UK, and 300 million people worldwide. There are an estimated 600,000 emergency admissions and 1,200 deaths in the UK annually11. Asthma is characterised by airway hyper-responsiveness, airway inflammation, variable airway expiratory limitation, hypersecretion of mucus, and airway remodelling12.
Numerous cell types in the mammalian lung express β2-AR, including airway smooth muscle, and epithelial and inflammatory cells. The aim of managing asthma is to reverse or prevent acute bronchoconstriction and inflammation, associated with β2-AR signalling.
Asthma symptoms are controlled in most asthmatics by short-acting β2-AR agonists (SABA), inhaled corticosteroids and long-acting β2-AR agonists (LABA). However, long-term use of LABA alone can lead to tolerance and increased risk of asthma exacerbation and even death13. Tolerance correlates with down-regulation of the β2-AR in the lungs of rodents14 and on circulating lymphocytes from asthmatics15.
Acute use of β-AR antagonists, regardless of their selectivity for the β1-AR and β2-AR, can cause bronchoconstriction in patients with asthma and, as in patients with CHF, have been contraindicated since the 1960s. Bronchoconstriction is thought to be due to blockade of the effects of endogenous epinephrine and/or inactivation of constitutively active β2-AR. Nevertheless, in recent years, there’s been much interest in the use of β-blockers in the management of patients with asthma. In non-clinical models of asthma and in clinical studies, different β-blockers have had very different effects.
In a murine model of antigen-driven airway inflammation and hyper-responsiveness, acute administration of the non-selective β-blockers nadolol or carvedilol increased airway resistance, whereas chronic administration for 28 days reduced airway resistance, which correlated with an increase in β-AR density in the lung14. In further studies in the allergen-sensitised mouse model, chronic dosing with the β-blockers nadolol (non-selective) or ICI 118,551 (β2-AR selective) reduced airway hypersensitivity, inflammation, and mucin content, and reversed pathological changes in the airway epithelium16,17.
Because of those unexpected results in murine models of asthma, two small open-label, pilot studies of rising doses of the non-selective β-blocker nadolol were done in patients with mild-to-moderate asthma in a USA centre18,19. Nadolol was generally well tolerated, and chronic administration led to a significant, dose-dependent reduction in airway hypersensitivity to inhaled methacholine, but a dose-independent 5% reduction in FEV1. A multicentre, placebo-controlled study of nadolol is in progress (ClinicalTrials.gov). On the other hand, a recent placebo-controlled trial of propranolol, another non-selective β-blocker, administered with concomitant ICS and tiotropium bromide (an anticholinergic bronchodilator), had no effect on airway hypersensitivity and caused a small but statistically insignificant fall in FEV1 in patients with mild-to-moderate asthma20. Nadolol, propranolol, carvedilol and ICI 118,551 all antagonise the β2-AR to some extent.
Their different effects of the β-blockers on hypersensitivity and FEV1 in the murine models and clinical studies have now been shown to be a result of ligand bias: preferential activation of one pathway over another by a ligand. The differences reflect their capacity to modulate binding of the scaffold protein, β-arrestin, to the β2-AR21 (Figure 2). Binding of β-arrestin activates extracellular regulated kinase (ERK) signalling, which is thought to be detrimental in asthma. Nadolol and ICI 118,551 turn off ERK signalling, while propranolol and carvedilol are partial agonists. Therefore, nadolol and ICI 118,551 have broncho-protective effects, while propranolol and carvedilol do not22.
Our first study of TR4 in patients with asthma is in progress.
Figure 2. Regulation of β2-AR by beta arrestin
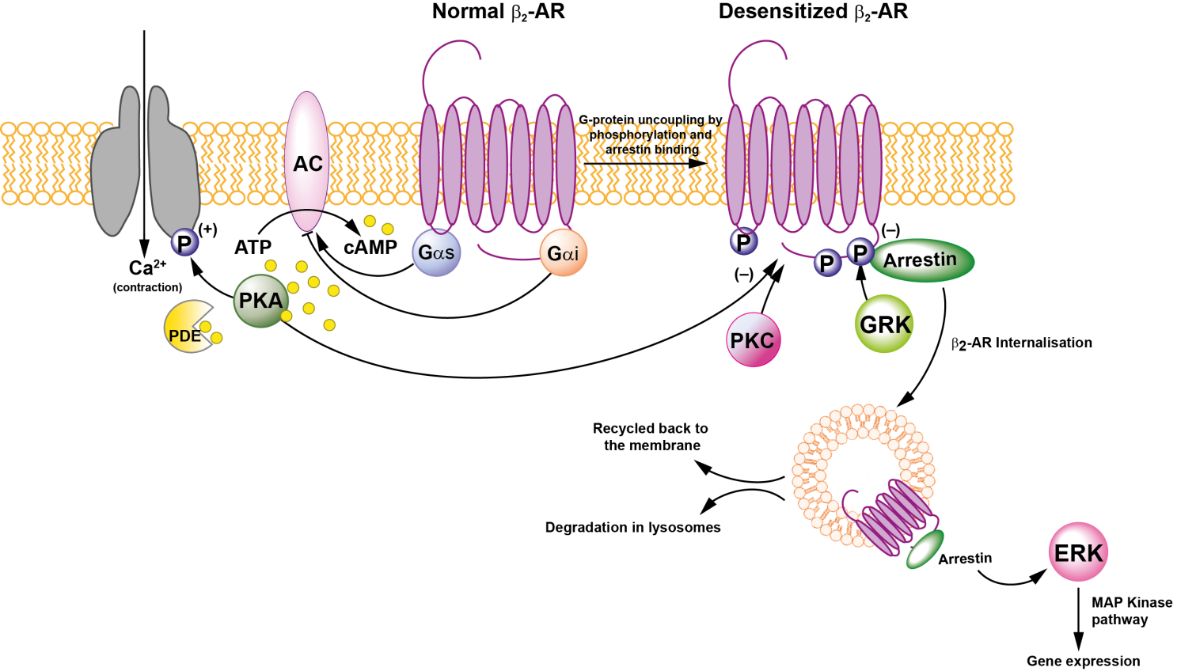
AC, adenylate cyclase; arrestin, β-arrestin; β2-AR, beta-2 adrenoceptor; Ca++, calcium; cAMP, cyclic adenosine monophosphate; ERK, extracellular regulated kinase; αs, heterotrimeric G-protein alpha subunit; Gαi, heterotrimeric G‑protein inhibitory subunit; GRK, G-protein receptor kinase; MAP kinase, mitogen-activated protein kinase; PDE, phosphodiesterase IV/3B; PKA, protein kinase A; (+), increases/activates, (-), decreases/inhibits; P, phosphorylation.
Pancreatic cancer
Pancreatic cancer is the 11th most common cancer in the UK, and accounts for 3% of all cancers. About 96% of pancreatic cancers are pancreatic ductal adenocarcinoma (PDAC), and theIR incidence is increasing23,24. PDAC arises from cells of the pancreatic duct or ductules through which digestive enzymes and bicarbonate reach the small intestine.
60-80% of PDAC arise from the head of the pancreas, which contains the common bile duct (Figure 3). Patients with obstruction of the common bile duct develop jaundice and present earlier than patients with tumours of the body and tail of the pancreas, which do not cause jaundice and present with weight loss and/or pain. Tumours are staged on the basis of their size, lymph node status and metastasis (“TNM”). Prognosis depends on tumour stage, but the prognosis is poor – the median survival is 6 months. Only 10-20% of patients have tumours that are amenable to surgical resection at presentation. The poor prognosis is due to the late onset of symptoms, aggressive local invasion, metastases, and the weak response to chemotherapy. Chemotherapy with gemcitabine, either as adjuvant therapy post-operatively or as standard of care, offers only a modest benefit.
Figure 3. Pancreas, pancreatic duct and common bile duct

There’s growing evidence from pre-clinical studies that β2-AR signalling regulates multiple cellular processes that contribute to initiation and progression of cancer, including inflammation, angiogenesis, apoptosis and cell motility and trafficking25,26. Human pancreatic cell lines express both β1-AR and β2-AR, but β2-AR expression is about two-fold higher than that of β1-AR. ICI 118,551, a selective β2-AR blocker, and propranolol, a non-selective β1-AR and β2-AR blocker, significantly suppressed cell invasion and proliferation compared to control or metoprolol, a selective β1-AR blocker27. The β2-AR blockers also inhibited expression of: CREB1 (CAMP Responsive Element Binding Protein 1), a protein coding gene; NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells), a protein complex that controls transcription of DNA, cytokine production and cell survival; and activator protein (AP-1), a protein that increases gene transcription. Furthermore, the β2-AR blockers downregulated the expression of the following target genes MMP-9 and MMP-2 (matrix metalloproteinase), which degrade extracellular matrix as a prerequisite for cellular invasion; VEGF (vascular endothelial growth factor, a protein that stimulates blood vessel formation); and COX-2 (cyclo-oxygenase). β2-AR blockers suppressed cell invasion via inhibition of both classical and non-classical pathways, whereas the β1-AR blocker inhibited only the classical pathway.
ICI 118,551 inhibited cell proliferation and potentiated the effect of gemcitabine on apoptosis in human pancreatic cell lines, probably by activation of NF-κB and expression of its targets28.
Compared with the normal pancreas, the density and size of nerves and expression of norepinephrine is high in PDAC. Recently, Renz et al (2018) showed that in genetically engineered KPC mice29, catecholamines promoted β2-AR-dependent PDAC development, nerve growth factor (NGF) secretion, pancreatic nerve density, and phosphorylation of ERK (extracellular signal-related kinase) and CREB (extracellular signal-related kinase)30 (Figure 4). Compared with gemcitabine alone, addition of ICI 118,551 to gemcitabine reduced NGF expression, reduced nerve density and increased survival (Figure 5). Also, analysis of PDAC patient cohorts showed a correlation between reduced neurotrophic expression, reduced nerve density, and increased survival in PDAC patients taking non-selective β-blockers . Those findings suggest that catecholamines drive β2-AR dependent upregulation of neurotrophins, which in turn increase sympathetic innervation and local norepinephrine accumulation.
Figure 4. The β-agonist isoproterenol and the catecholamine norepinephrine increase phosphorylation of ERK and CREB in a genetically engineered KPC mouse model of PDAC, whereas ICI 118,551 inhibits the responses
")
Figure 5. Addition of ICI 118,551 to gemcitabine in a genetically engineered KPC mouse model of PDAC: A. increases survival; and B. reduces tumour volume

An earlier study also showed that patient survival is worse in the 70% of PDAC patients with perineural invasion in their biopsies31. A meta-analysis of clinical trials of adjuvant treatments compared with gemcitabine alone in patients with resected pancreatic cancer showed significant survival only for fluoropyrimidine, an oral chemotherapeutic agent32.
In summary, there’s an unmet need for medical treatments of PDAC. There’s good evidence to justify a study of TR4 as add-on therapy to gemcitabine in patients with PDAC who are considered unsuitable for surgery and whose tumour is ideally at an early stage. Such tumours are probably more amenable to medical treatment 32. Potential outcome measures are: survival; blood biomarkers27,29 methylation status of ADAMTS1 and BNC1 genes33; and tumour biomarkers CREB1, NF-κB, AP-1, MMP-9, MMP-2; VEGF, and COX-2 in biopsies taken by endoscopic retrograde cholangio-pancreatography34 (ERCP).
References
- Black J, Fitzgerald D. Evolving concepts concerning cardiac β-adrenoceptor function in heart failure. Curr Pharm Des 2010; 16: 4148–4158.
- Baker AJ. Adrenergic signaling in heart failure: a balance of toxic and protective effects. Pflugers Arch 2014; 466: 1139–1150.
- Lands AM, Arnold A, McAuliff JP, Luduena FP, Brown JT. Differentiation of receptor systems activated by sympathomimetic amines. Nature 1967; 214: 597–598.
- Bilski AJ, Halliday SE, Fitzgerald JD, Wale JL. The pharmacology of a [beta] 2-selective adrenoceptoaAntagonist (ICI 118,551). J Cardiovasc Pharmacol 1983; 5: 430–437.
- Baker JG. The selectivity of beta-adrenoceptor antagonists at the human beta1, beta2 and beta3 adrenoceptors. Br J Pharmacol 2005; 144: 317–322.
- Waagstein F, Hjalmarson A, Varnauskas E, Wallentin I. Effect of chronic beta-adrenergic receptor blockade in congestive cardiomyopathy. Br Heart J 1975; 37: 1022–1036.
- Chatterjee S, Biondi-Zoccai G, Abbate A, D’Ascenzo F, Castagno D, Van Tassell B, Mukherjee D, Lichstein E. Benefits of β blockers in patients with heart failure and reduced ejection fraction: network meta-analysis. BMJ 2013; 346: f55 doi: 10.1136/bmj.f55.
- Metra M, Nodari S, Bordonali T, Milani P, Fracassi F, Dei Cas L. β-blocker therapy of heart failure: an update. Exp Opin Pharmacother 2007; 8: 289–298.
- Hasseldine AR, Harper EA, Black JW. Cardiac-specific overexpression of human beta 2 adrenoceptors in mice exposes coupling to both Gs and Gi proteins. Br J Pharmacol 2003; 138: 1358–1366.
- Molenaar P, Christ T, Berk E, Engel A, Gillette KT, Galindo-Tovar A, Ravens U, Kaumann AJ. Carvedilol induces greater control of β2-than β1-adrenoceptor-mediated inotropic and lusitropic effects by PDE3, while PDE4 has no effect in human failing myocardium. Naunyn Schmiedeberg’s Arch Pharmacol 2014; 387: 629–640.
- Poole-Wilson PA, Swedberg K, Cleland JG, Di Lenarda A, Hanrath P, Komajda M, Lubsen J, Lutiger B, Metra M, Remme WJ, Torp-Pedersen C. Comparison of carvedilol and metoprolol on clinical outcomes in patients with chronic heart failure in the Carvedilol or Metoprolol European Trial (COMET): randomised controlled trial. Lancet 2003; 362: 7–13.
- Asthma UK. Asthma facts and statistics https://www.asthma.org.uk/about/media/facts-and-statistics/: Asthma UK; 2016 [cited 2016 18 July 2016].
- GINA. Global Initiative for Asthma. Global Strategy for Asthma Management and Prevention, 2016. Available from www.ginasthma.org. 2016.
- Salpeter SR, Buckley NS, Ormiston TM, Salpeter EE. Meta-analysis: effect of long-acting beta-agonists on severe asthma exacerbations and asthma-related deaths. Ann Intern Med. 2006; 144: 904–912.
- Callaerts-Vegh Z, Evans KL, Dudekula N, Cuba D, Knoll BJ, Callaerts PF, Giles H, Shardonofsky FR, Bond RA. Effects of acute and chronic administration of β-adrenoceptor ligands on airway function in a murine model of asthma. Proc Natl Acad Sci USA 2004; 101: 4948–4953.
- Aziz I, Lipworth BJ. A bolus of inhaled budesonide rapidly reverses airway subsensitivity and beta2-adrenoceptor down-regulation after regular inhaled formoterol. Chest 1999; 115: 623–8.
- Nguyen LP, Omoluabi O, Parra S, Frieske JM, Clement C, Ammar-Aouchiche Z, et al. Chronic Exposure to Beta-Blockers Attenuates Inflammation and Mucin Content in a Murine Asthma Model. Am J Respir Cell Mol Biol. 2008;38(3):256–262.
- Lin R, Peng H, Nguyen LP, Dudekula NB, Shardonofsky F, Knoll BJ, Parra S, Bond RA. Changes in β2-adrenoceptor and other signalling proteins produced by chronic administration of ‘β-blockers’ in a murine asthma model. Pul Pharmacol Ther 2008; 21: 115–124.
- Hanania NA, Singh S, El-Wali R, Flashner M, Franklin AE, Garner WJ, Dickey BF, Parra S, Ruoss S, Shardonofsky F, O’Connor BJ. The safety and effects of the beta-blocker, nadolol, in mild asthma: an open-label pilot study. Pulm Pharmacol Ther 2008; 21: 134–41.
- Hanania NA, Mannava B, Franklin AE, Lipworth BJ, Williamson PA, Garner WJ, Dickey BF, Bond RA. Response to salbutamol in patients with mild asthma treated with nadolol. Eur Respir J 2010; 36: 963–965.
- Short PM, Williamson PA, Anderson WJ, Lipworth BJ. Randomized placebo-controlled trial to evaluate chronic dosing effects of propranolol in asthma. Am J Respir Crit Care Med 2013; 187: 1308–1314.
- Walker JKL, Penn RB, Hanania NA, Dickey BF, Bond RA. New perspectives regarding β2-adrenoceptor ligands in the treatment of asthma. Br J Pharmacol 2011;163: 18–28.
- Stark A, Eibi G. Pancreatic ductal adenocarcinoma. doi: 10.3998/panc.2015.14.
- Rawls P, Sunkara T, Gaduputi V. Epidemiology of pancreatic cancer: global trends, etiology and risk factors. World J Oncol 2019; 10: 10–27.
- Cole SW, Sood AK. Molecular pathways: beta-adrenergic signaling in cancer. Clin Cancer Res 2012; 18: 1201–1206.
- Chang A, Kim-Fuchs C, Le CP, Hollande F, Sloan EK. Neural regulation of pancreatic cancer: a novel target for intervention. Cancers 2015; 7: 1292–312.
- Zhang D, Ma Q-Y, Hu H-T, Zhang M. β2-adrenergic antagonists suppress pancreatic cancer cell invasion by inhibiting CREB, NFκB and AP-1. Cancer Biol Ther 2010; 10: 19–29.
- Shan T, Ma Q, Zhang D, Guo K, Wang F, Wu E. β2-adrenoceptor blocker synergizes with gemcitabine to inhibit the proliferation of pancreatic cancer cells via apoptosis induction. Eur J Pharmacol 2011; 665: 1–7.
- Lee J, Komar C, Bengsch F, Graham K, Beatty G. Genetically engineered mouse models of pancreatic cancer: KPC model (LSL-KrasG12D/+; LSL-Trp53R172H/+; Pdx-1-Cre), its variants and their application in immune-oncology drug discovery. Curr Protoc Pharmacol 2007; 73: 1–14.
- Renz BW, Takahashi R, Tanaka T, Macchini M, Hayakawa Y, Dantes Z, Maurer HC, Chen X, Jiang Z, Westphalen CB, Ilmer M, Valenti G, Mohanta SK, Habenicht AJR, Middelhoff M, Chu T, Nagar K, Tailor Y, Casadei R, Di Marco M, Kleespies A, Friedman RA, Remotti H, Reichert M, Worthley DL, Neumann J, Werner J, Iuga AC, Olive KP, Wang TC. β2-adrenergic neurotrophin feedforward loop promotes pancreatic cancer. Cancer Cell 2018; 34: 863–867.
- Stark AP, Sacks GD, Rochefort MM, Donahue TR, Reber HA, Tomlinson JS, Dawson DW, Eibl G, Hines OJ. Long-term survival in patients with pancreatic ductal adenocarcinoma. Surgery 2016; 159: 1520–1527.
- Chen H, He R, Shi X, Zhou M, Zhang H, Qin R. Meta-analysis on resected pancreatic cancer: a comparison between adjuvant treatments and gemcitabine alone. BMC Cancer 2018; 18: 10034.
- Eissa MAL, Lerner L, Abdelfatah E, Shankar N, Canner JK, Hasan NM, Yaghoobi V, Huang B, Kerner Z, Takaesu F, Wolfgang C, Kwak R, Ruiz M1, Tam M, Pisanic TR, Iacobuzio-Donahue CA, Hruban RH, He J, Wang TH, Wood LD, Sharma A, Ahuja N. Promoter methylation of ADAMTS1 and BNC1 as potential biomarkers for early detection of pancreatic cancer in blood. Clin Epigenetics 2019; 11: 59.
- https://www.hey.nhs.uk/patient-leaflet/ercp-endoscopic-retrograde-cholangiopancreatography/